Molekulare Simulationen: Von biomolekularen Strukturen zur Funktion
Forschungsbericht (importiert) 2014 - Max-Planck-Institut für Biophysik
Molekulare Simulation – ein Werkzeug zur Untersuchung biomolekularer Prozesse
Die Zellen lebender Organismen bestehen aus Proteinen, Nukleinsäuren und Lipiden. Die Struktur, Dynamik und Wechselwirkung dieser Moleküle bestimmen wesentlich die Mechanismen biologischer Prozesse. Um molekulare Strukturen und assoziierte dynamische Prozesse zu untersuchen, wurde ein breites Spektrum experimenteller Methoden entwickelt, von der biochemischen Charakterisierung hin zur detaillierten Bestimmung atomarer Positionen in der Röntgenkristallographie. Diese experimentellen Methoden werden zunehmend durch theoretische Beschreibungen ergänzt, die in wachsendem Maße als Partner des Experiments akzeptiert werden [1, 2]. Moderne molekulare Simulationsmethoden erlauben es, biomolekulare Systeme mit oft mehreren Millionen Atomen über Zeiträume von Femtosekunden hin zum Millisekunden-Bereich zu untersuchen.
In der Molekulardynamik-Simulation wird die thermische Bewegung jedes einzelnen Atoms verfolgt, indem deren gekoppelte Newton‘sche Bewegungsgleichungen numerisch gelöst werden. Dabei werden die auf die Atome wirkenden Kräfte entsprechend einer quantenmechanischen oder einer ihr angenäherten klassischen Potenzialenergiefläche berechnet. Mit dieser detaillierten Beschreibung kann man Proteinen und anderen Biomolekülen sozusagen bei der Arbeit zuschauen. Wie im Folgenden dargelegt, kann man mit Simulationen komplexe Experimente interpretieren und so scheinbare Widersprüche zwischen unterschiedlichen Messungen und Interpretationen auflösen [3], entscheidende Lücken in der experimentellen Beschreibung füllen [4], oder – gleichsam wie in einem Experiment – neue molekulare Prozesse entdecken [5].
Experimente in scheinbarem Widerspruch: Pikosekunden-Dynamik im Lichtschalter PYP
Photoactive Yellow Protein (PYP) ist ein Lichtschalter-Protein, in dem die Absorption eines Photons innerhalb von Pikosekunden eine starke Änderung der lokalen Struktur eines kovalent gebundenen Farbstoffs induziert. Die daraus resultierende mechanische Spannung wird über eine Kaskade von Strukturänderungen bis hin zu einer teilweisen Entfaltung des Proteins im Millisekunden-Bereich abgebaut. Diese Strukturänderungen konnten mit erstaunlichem Detail durch zeitaufgelöste Kristallographie verfolgt werden [6], wobei es aber interpretatorisch zu einem entscheidenden Unterschied, verglichen mit einer anderen Messung, kam [3]: In einer Röntgenstruktur war nach 100 Pikosekunden der für die gesamte Dynamik entscheidende Torsionswinkel des gebundenen Farbstoffs schon vom trans- in den cis-Zustand isomerisiert; in der von einer anderen Gruppe bestimmten Struktur wurde zu diesem Zeitpunkt jedoch ein Übergangszustand mit einem völlig anderen Torsionswinkel beobachtet. Diese unterschiedlichen Ergebnisse würden zwei sehr unterschiedliche Interpretationen des Zusammenhangs zwischen Photochemie und Proteindynamik bedingen, ausgehend von einem lokal schon relaxierten Grundzustand oder von einer Struktur, die noch dem optisch erzeugten angeregten Zustand entspricht.
Quantenchemische Berechnungen und molekulare Simulationen haben es möglich gemacht, diese Diskrepanz auszuräumen. Abbildung 1 verdeutlicht die Schwierigkeit, den Torsionswinkel der Doppelbindung des Farbstoffs allein aus der experimentell beobachteten Elektronendichte zu bestimmen. Selbst mit hoher kristallographischer Auflösung ist es schwierig, den Torsionswinkel des Farbstoffs genau abzuschätzen, da dieser durch kleine, aber korrelierte Verschiebungen der atomaren Positionen stark abgeändert werden kann.
 mit atomarer Struktur des darin gebundenen Farbstoffs, 100 Pikosekunden nach Lichtabsorption (rechts; vgl. [3, 6]). Quantenchemische Berechnungen und molekulare Simulationen zeigen, dass nur die cis-Struktur (grün) energetisch möglich ist, nicht aber ein Übergangszustand (rot), obwohl beide Strukturen nahezu gleich gut in die hochaufgelöste Elektronendichte passen (vgl. [3]).")
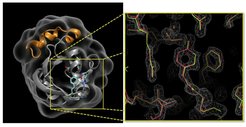
Abb. 1: Lichtsensor-Protein PYP (Photoactive Yellow Protein, links) mit atomarer Struktur des darin gebundenen Farbstoffs, 100 Pikosekunden nach Lichtabsorption (rechts; vgl. [3, 6]). Quantenchemische Berechnungen und molekulare Simulationen zeigen, dass nur die cis-Struktur (grün) energetisch möglich ist, nicht aber ein Übergangszustand (rot), obwohl beide Strukturen nahezu gleich gut in die hochaufgelöste Elektronendichte passen (vgl. [3]).
Quantenchemische Berechnungen mittels der Dichtefunktionalmethode und sogenannte QM/MM Simulationen, in denen ein Teil des Systems quantenmechanisch und der Rest klassisch behandelt werden, ergaben aber ein klares Resultat: Nur ein cis-Winkel ist energetisch möglich. Übergangszustandsartige Anfangsstrukturen relaxieren deshalb rasch in den cis-Bereich. Aufgrund der Berechnungen ist somit klar, dass die lichtinduzierte, für die weitere Zeitentwicklung entscheidende lokale Strukturänderung schon nach wenigen Pikosekunden abgeschlossen ist. Die verbleibenden, größerskaligen Umordnungen, bis hin zur Teilentfaltung, sind damit durch dieses erste, schnelle Ereignis bedingt.
Über das Experiment hinaus – atomar aufgelöste Dynamik in der enzymatischen Katalyse
Moderne molekulare Simulationen erlauben es auch, direkt den Übergang zwischen Anfangs- und Endprodukten einer enzymatischen Reaktion zu untersuchen. Auf den ersten Blick mag dies unmöglich erscheinen, da die Reaktionszeiten etwa für Ribonuklease H1 im Bereich von Sekunden liegen [4], quantenmechanische Simulationen aber nur den Piko- bis Nanosekundenbereich abdecken. Allerdings kann man mit modernen Algorithmen selektiv den Bereich des chemischen Übergangs untersuchen.
So konnte beispielsweise gezeigt werden, wie Ribonuklease H1 das RNA-Rückgrat in einer RNA:DNA-Hybridstruktur schneidet [7]; ein Prozess, der sowohl für die Replikation von DNA als auch für den Einbau viraler Genome, zum Beispiel in HIV-infizierten Zellen, von entscheidender Bedeutung ist. Noch offen war die Frage, warum Magnesiumionen (Mg2+) in diesem bei vielen zellulären Prozessen auftretenden Zwei-Ionen-Mechanismus, nicht aber die biologisch ebenso verbreiteten Kalziumionen (Ca2+) katalytisch aktiv sind. Die Berechnungen zeigten, dass Ca2+ in der Tat zu einer dramatischen Reduktion der kinetischen Rate führt, wie dies auch im Experiment beobachtet werden konnte [4]; und zudem kann man in der Simulation die Ursachen dieses Effekts sogar direkt erkunden. Wider Erwarten, so stellte sich heraus, ist es weniger eine Veränderung der lokalen Struktur der Ionen- und RNA-Bindungsstelle, sondern primär ein im Grunde einfacher, elektrostatischer Effekt, der zur Bevorzugung der Magnesiumionen führt: Der entscheidende Hydrolyseschritt verläuft mit Kalziumionen nämlich sehr langsam, da diese im Vergleich zu Magnesium größeren Ionen das für die Spaltung unabdingbare Wassermolekül nicht effizient durch Abziehen von Elektronenladung aktivieren können - was mithilfe von Magnesiumionen wiederum gelingt.
Damit konnte dank Simulationen gezeigt werden, warum Ca2+ bei der Spaltung von RNA inaktiv ist. Durch ein Experiment allein wäre dies, wenn überhaupt, nur sehr bedingt möglich gewesen, da die eigentlichen Reaktionsübergänge sehr rasch, die Wartezeiten zwischen den Übergängen von Reaktanden zu Produkten jedoch um viele Größenordnungen länger sind.
Ins Unbekannte vorstoßen – biomolekulare Simulation als Computerexperiment
Molekulare Simulationen können auch als Werkzeug für Entdeckungen neuer Prozesse dienen. Ähnlich einem Experiment werden zunächst bestimmte Anfangszustände präpariert und dann die sich ergebende zeitliche Entwicklung des Systems betrachtet. Die beobachteten Ereignisse helfen dabei, neue Hypothesen zu formulieren, die dann experimentell oder durch weitere Berechnungen getestet werden können.
Im Falle des Komplex I, einer für die effiziente biologische Energieumwandlung entscheidenden Protonenpumpe innerhalb der Atmungskette von Bakterien und Mitochondrien, haben klassische Molekulardynamik-Simulationen eine dramatische Änderung in der Struktur des im Protein eindringenden Wassers gezeigt. Es bildete sich insbesondere nach mehreren Hundert Nanosekunden eine lineare Kette eindimensional angeordneter Wassermoleküle, die die Oberfläche mit dem Inneren des Proteins verbindet (Abb. 2; [5]). Diese Kette wird von Wasserstoffbrücken zusammengehalten und endet an einem negativ geladenen Glutamat, das in der Gensequenz des Komplex I vieler Organismen erhalten und konserviert ist. Weitere Berechnungen wiederum lassen eine wichtige Rolle sowohl der Wasserkette und demnach auch des Glutamats bei der Funktionsweise von Komplex I vermuten: Quantenmechanische (QM/MM) Simulationen zeigten, dass die Kette als sehr effizienter, eindimensionaler Protonenleiter fungiert. Innerhalb von Pikosekunden kann so ein Proton aus der Lösung tief in den Membranbereich eindringen - einem für die biologische Energieumwandlung durch Komplex I insgesamt entscheidenden Ereignis.
 mit einer Vergrößerung (rechts), die eine in der Simulation spontan gebildete, eindimensionale Wasserkette zeigt (blauer Pfeil; vgl. [5]). Weitere Simulationen zeigten, dass diese Kette ein hervorragender Protonenleiter ist. Das Auflösen der Kette nach dem Protonentransfer hilft, den graduellen Abbau des Protonengradienten durch die Membran zu verhindern, sodass dieser Gradient nachfolgend für die Gewinnung gespeicherter Energie mittels ATP-Bildung genutzt werden kann.")
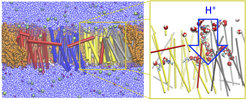
Der ebenfalls in der Simulation beobachtete Zerfall der Wasserkette nach dem erfolgreichen Protonentransfer, bedingt durch das dadurch veränderte elektrische Feld, ist ebenfalls von großer Bedeutung für die Energieeffizienz: So kann nämlich verhindert werden, dass durch den Membranbereich gepumpte Protonen leicht und einfach wieder zurückdiffundieren können.
Ausblick
Durch rasch fortschreitende Entwicklungen neuer Rechnersysteme, verbesserter Algorithmen und optimierter Beschreibungen ist es zunehmend möglich, selbst komplexe biomolekulare Prozesse durch realistische molekulare Simulationen zu untersuchen [1]. Anwendungen molekularer Simulationen reichen von schnellen Reaktionsprozessen bis hin zur Strukturbildung auf zellulären Skalen. Somit tragen Simulationen wesentlich zu einem Verständnis der dem Leben zugrunde liegenden biomolekularen Prozesse bei und bilden damit eine wichtige Basis auch für Anwendungen in Medizin und Biotechnologie.
Literaturhinweise
Journal of the American Chemical Society 133, 8934-8941 (2011)